
Why is it that once the protein structure of one member of a family has been solved
ab initio, it is easier to solve more members of the same family? Compare the
structures of two members of the crystallin family which have 90% sequence identity.
The biggest sequence difference is a deletion of 1 residue (residue 86 in red) in the
linker connecting the two domains in
(at the bottom of the molecule in the display)
When identical or similar structures exist in different crystallographic environments,
similarities between their diffraction patterns, which are direcly related to their Fourier
transforms, would be expected. The technique of Molecular Replacement
exploits this similarity to determine phases. The dominant application is
The rigid bodies are described by the coordinates:
x1 unknown, observed or target
x2 known or model
The relationships are described by the operations:
R rotation matrix expressed in 3 Eulerian angles
t translation vector tx ty tz
Molecular replacement is a six-dimensional problem with order 6, or 6 N in case of N molecules in the
asymmetric unit.
Examine the film to see how the rotation and translation steps relate the model
x1
(e.g.
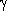 D Crystallin
compared to B Crystallin. A structural
difference is that the C-terminal tails have a different conformation. Yet,
it is clear that the three-dimensional structures of both proteins are
at first sight indistinguishable. Proteins have similar three-dimensional
conformations for sequence identities as low as 20%. This phenomenon
is at the basis of molecular replacement.
D Crystallin
compared to B Crystallin. A structural
difference is that the C-terminal tails have a different conformation. Yet,
it is clear that the three-dimensional structures of both proteins are
at first sight indistinguishable. Proteins have similar three-dimensional
conformations for sequence identities as low as 20%. This phenomenon
is at the basis of molecular replacement.
The principle of molecular replacement
To express the relationship between 2 identical rigid bodies, 6 parameters,
3 rotational and 3 translational, are required.
 or
or  1 2
3
B Crystallin of which the structure was known)
and the target
x2
(e.g. D Crystallin before that
its structure was known).
1 2
3
B Crystallin of which the structure was known)
and the target
x2
(e.g. D Crystallin before that
its structure was known).
![]() Animated GIF "Movie" "Very High" Speed
Animated GIF "Movie" "Very High" Speed![]() Animated GIF "Movie" "High" Speed
Animated GIF "Movie" "High" Speed![]() Animated GIF "Movie" "Slow" Speed
Animated GIF "Movie" "Slow" Speed![]() Animated GIF "Movie" "Very Slow" Speed
Animated GIF "Movie" "Very Slow" Speed
![]() Welcome Page
Welcome Page
![]() Course Information
Course Information
![]() Course Index
Course Index
Copyright © 1995-2007 Birkbeck College